
14.6.1 POJA-L6184+6185 Immunostaining for dystrophin in Duchenne muscular dystrophy (DMD)
14.6.1 POJA-L6184+6185 Immunostaining for dystrophin in Duchenne muscular dystrophy (DMD)
14.6.1 POJA-L6184+6185 Immunostaining for dystrophin in Duchenne muscular dystrophy (DMD)
(By courtesy of H. ter Laak PhD Section Neuropathology, retired staff member Department of Pathology, Radboud university medical center, Nijmegen, The Netherlands)
Title: Immunostaining for dystrophin in Duchenne muscular dystrophy (DMD)
Description:
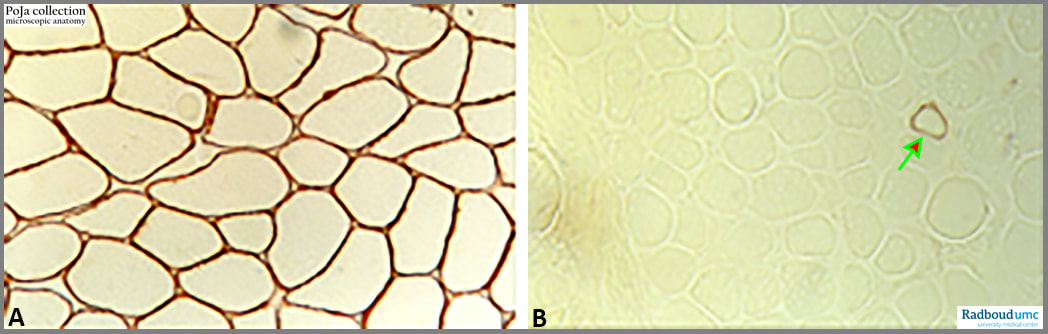
Duchenne muscular dystrophy (DMD) with absence of dystrophin, human.
(A): Normal skeletal muscle immunostained for the presence of dystrophin.
(B): Duchenne patient showing absence of dystrophin, except in one fibre (arrow). The only single reactive myofibre is a revertant fibre that recovered from the reading frame and has mutated to normal expression.
Introduction:
Duchenne muscular dystrophy (DMD) is the most common hereditary neuromuscular disease. Mutations in the dystrophin gene lead to progressive muscle fibre degeneration and weakness. Cardiac and orthopaedic complications are common, and death usually occurs in the twenties due to respiratory muscle weakness or cardiomyopathy.
Aetiology: DMD is a genetic disease due to the mutation of the dystrophin gene, located on chromosome Xp21. It is inherited as an X- linked recessive trait. Mutations in the dystrophin gene result in so-called dystrophinopathies (a.o. Duchenne muscular dystrophy, Becker muscular dystrophy). Mutations result in a limited production of the dystrophin protein, which results in loss of the myofibre membrane integrity with repeated cycles of necrosis and regeneration. Fibrous connective tissue and fat progressively replace muscle leading to clinical features.
Epidemiology: as DMD is inherited as an X-linked recessive manner, boys are more frequently affected than girls.
Pathophysiology: Dystrophin is a large cytoskeletal protein that facilitates interactions between the cytoskeleton, cell membrane, and extracellular matrix. It is located at the inner site of the sarcolemma in muscle tissues. Dystrophin is a part of the dystrophin-glycoprotein complex (DGC), which plays an important role as being a structural unit of muscle. In DMD, both dystrophin and DGC proteins are missing, leading to excessive membrane fragility and permeability resulting in dysregulation of e.g., calcium homeostasis, oxidative damage. These factors play a crucial role in muscle cell necrosis followed by an elevated creatine kinase blood level. With ageing, the regenerative capacity of the muscles appears to be exhausted, and connective and adipose tissue gradually replace muscle fibres.
Histopathology: A muscle biopsy will demonstrate connective tissue proliferation (endomysial and perimysial fibrosis), scattered degeneration and regeneration of myofibres, resulting in a great variety in fibre sizes (atrophy and hypertrophy). Muscle fibre necrosis with mononuclear cell infiltrates, and replacement of muscle with adipose tissue and fat are present.
Keywords/Mesh: locomotor system, skeletal muscle, striated muscle, neuromuscular disease, sarcolemma, dystrophin, DMD, revertant fibre, pathology, POJA-collection
Title: Immunostaining for dystrophin in Duchenne muscular dystrophy (DMD)
Description:
Duchenne muscular dystrophy (DMD) with absence of dystrophin, human.
(A): Normal skeletal muscle immunostained for the presence of dystrophin.
(B): Duchenne patient showing absence of dystrophin, except in one fibre (arrow). The only single reactive myofibre is a revertant fibre that recovered from the reading frame and has mutated to normal expression.
Introduction:
Duchenne muscular dystrophy (DMD) is the most common hereditary neuromuscular disease. Mutations in the dystrophin gene lead to progressive muscle fibre degeneration and weakness. Cardiac and orthopaedic complications are common, and death usually occurs in the twenties due to respiratory muscle weakness or cardiomyopathy.
Aetiology: DMD is a genetic disease due to the mutation of the dystrophin gene, located on chromosome Xp21. It is inherited as an X- linked recessive trait. Mutations in the dystrophin gene result in so-called dystrophinopathies (a.o. Duchenne muscular dystrophy, Becker muscular dystrophy). Mutations result in a limited production of the dystrophin protein, which results in loss of the myofibre membrane integrity with repeated cycles of necrosis and regeneration. Fibrous connective tissue and fat progressively replace muscle leading to clinical features.
Epidemiology: as DMD is inherited as an X-linked recessive manner, boys are more frequently affected than girls.
Pathophysiology: Dystrophin is a large cytoskeletal protein that facilitates interactions between the cytoskeleton, cell membrane, and extracellular matrix. It is located at the inner site of the sarcolemma in muscle tissues. Dystrophin is a part of the dystrophin-glycoprotein complex (DGC), which plays an important role as being a structural unit of muscle. In DMD, both dystrophin and DGC proteins are missing, leading to excessive membrane fragility and permeability resulting in dysregulation of e.g., calcium homeostasis, oxidative damage. These factors play a crucial role in muscle cell necrosis followed by an elevated creatine kinase blood level. With ageing, the regenerative capacity of the muscles appears to be exhausted, and connective and adipose tissue gradually replace muscle fibres.
Histopathology: A muscle biopsy will demonstrate connective tissue proliferation (endomysial and perimysial fibrosis), scattered degeneration and regeneration of myofibres, resulting in a great variety in fibre sizes (atrophy and hypertrophy). Muscle fibre necrosis with mononuclear cell infiltrates, and replacement of muscle with adipose tissue and fat are present.
Keywords/Mesh: locomotor system, skeletal muscle, striated muscle, neuromuscular disease, sarcolemma, dystrophin, DMD, revertant fibre, pathology, POJA-collection